(1b) Chemistry of highly reactive diborane(4)
In the undergraduate class of chemistry, we study that borane (BH3) molecule easily dimerizes to form diborane(6) molecule (B2H6) in which two hydrogen atoms bridge two boron atoms through 3-center-2-electron bond. On the other hand, removal of two hydrogen atoms from diborane(6) gives diborane(4) molecule (B2H4) possessing B-B single bond. Among many reported diborane(4) molecules, bis(pinacholato)diborane(4), B2pin2, would be the most widely used molecule. Combination of this compound with transition metal catalyst enables to achieve diboration of alkynes and alkenes. With particular catalyst, C-H bonds of arenes and alkanes can also be borylated. However, examples of diborane(4) molecule without pπ-pπ stabilization from lone pairs of heteroatom substituents have been limited and their reactivity is not well-known.
We recently reported that diborane(4) having pinachol and two mesityl group showed the unique reactivity due to the existence of highly Lewis acidic boron center and reactive B-B single bond. An unsymmetrical diborane(4), pinB-BMes2, which could be prepared from the commercially available B2pin2 in one-step, directly reacted with CO to form a product incorporating two CO molecules through B-B bond cleavage. This unsymmetrical diborane(4) also reacted with 1 equiv. of tBu-isocyanide to afford cyclized boraindane derivative. Reaction with an excess amount of tBu-isocyanide gave isocyanide-coordinated boraalkene and its isomer. These reactions can be considered as the first example of C-N triple bond cleavage without transition metal at room temperature.
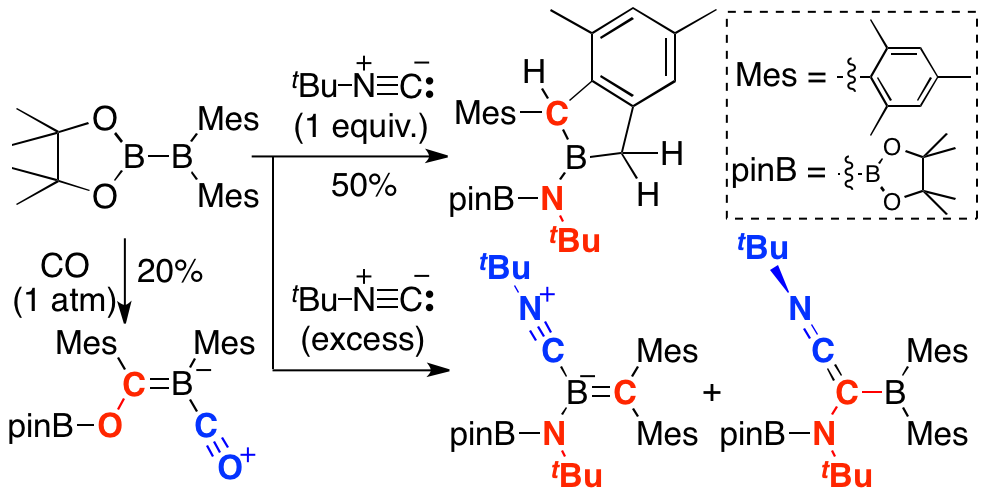
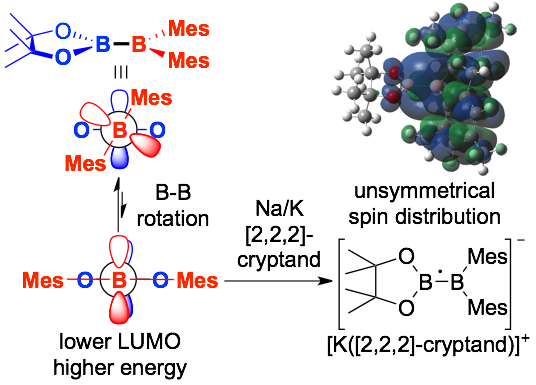
High reactivity of pinB-BMes2 could be attributed to the high Lewis acidity. In the presence of 2,2,2-cryptand, pinB-BMes2 was treated with Na/K alloy to generate radical anion derivative as deep purple crystals. DFT calcs revealed that the deep color related transitions involving SOMO. Combination of ESR spectroscopy and DFT calcs showed two boron atoms have different spin density reflecting the difference in Lewis acidity of these two boron atoms. Electrochemistry showed that the one-electron redction of pinB-BMes2 was easier than that of typical strong Lewis acid, BMes3. Theoretical calculations also revealed that rotation of B-B bond led to the overlapping two vacant p-orbitals to lower LUMO level for its strongly electron-accepting character.
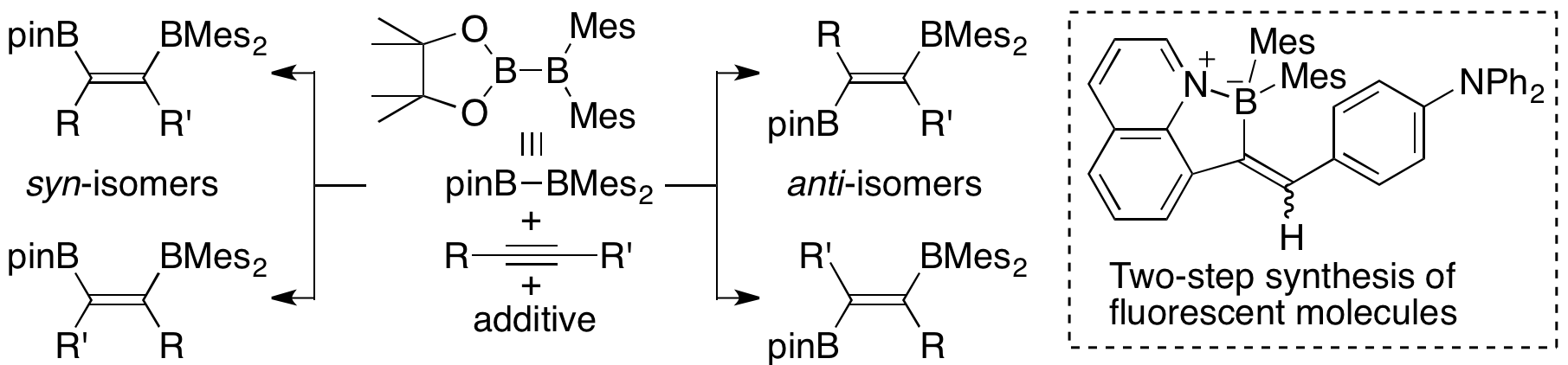
pinB-BMes2 could also react with alkyne in the absence of transition metal catalyst. Heating pinB-BMes2 with phenylacetylene gave an isomeric mixture of syn-diborylalkene. Addition of the catalytic amount of nBuLi reversed the isomeric ration of syn-diborylalkene. Addition of nBuLi and 1,2-dimethoxyethane (DME) led to a formation of anti-diborylalkene as the major product. Isolation of two possible reaction intermediates enabled us to estimate the reaction mechanism. By utilizing this reaction, we could synthesize quinoline derivatives showing emission in solid state in two-simple steps.
In the reaction of pinB-BMes2 with 2,6-dimethylphenylisocyanide (Xyl-NC), an interesting ring contraction was observed. pinB-BMes2 reacted with one equivalent of Xyl-NC to afford a spirocyclic compound having four-membered 1,2-oxaboretane through the ring contraction. DFT calcs revealed this reaction involved many rearrangement reactions. Usually Bpin is considered to be a stable substituent in organic chemistry, but this reaction showed the proximity of Bpin to highly Lewis acidic BMes2 moiety led to the ring contraction as a possible decomposition pathway.
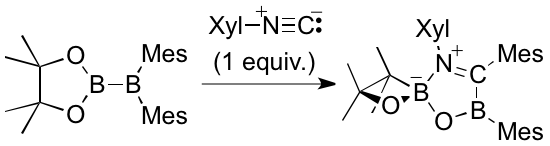
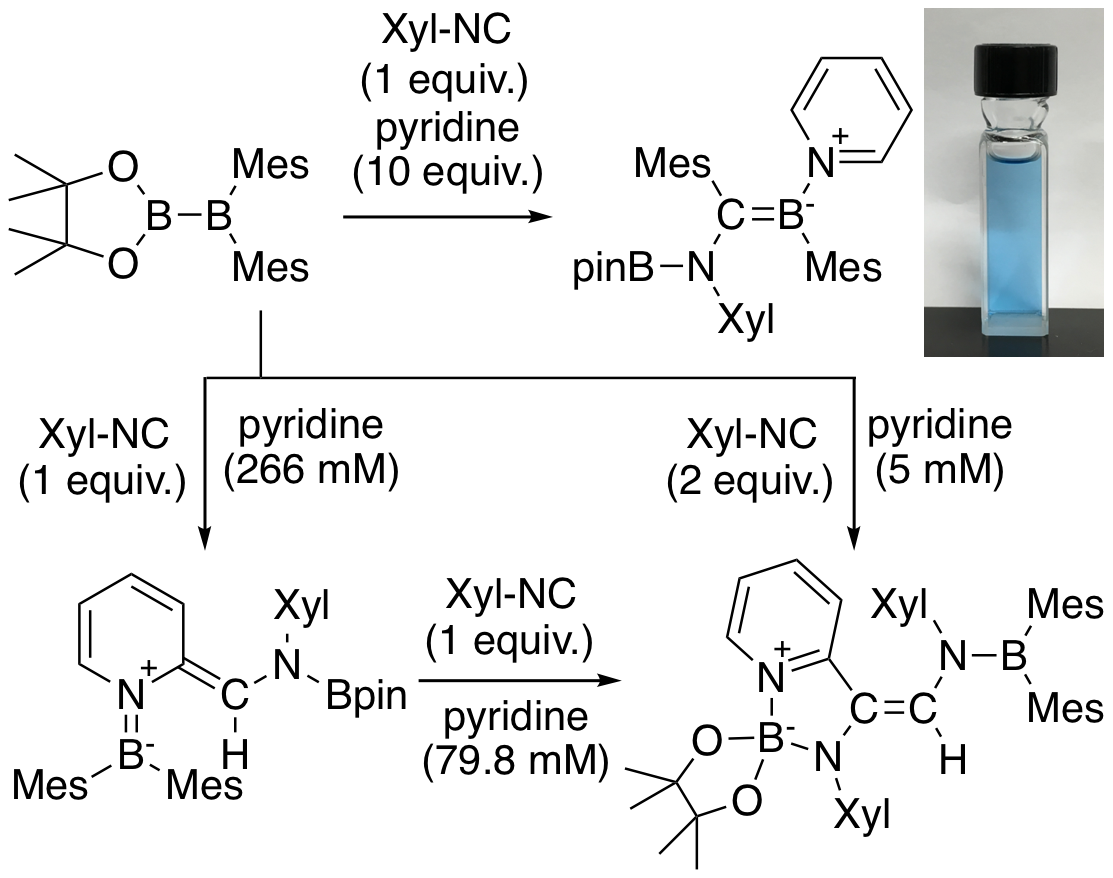
It was found that when pyridine was allowed to coexist in the previous reaction between pinB-BMes2 and isocyanide, an intense blue-colored pyridine-coordinated boraalkene was obtained. Blue organic compounds usually have a widely expanded π-conjugated structure, but this compound has only four 6-membered aromatic rings, being orthogonal to the Boraalkene plane with negligible π-conjugation. However, DFT calculations revealed that there is an intramolecular charge transfer transition from the boraalkene moiety to the coordinating pyridine ring, and the characteristic blue color is due to the unusual environment in which these two functional groups coexist. It was also clarified that when an excessive amount of pyridine reacted, the C-H bond at the 2-position of pyridine was functionalized with perfect selectivity, although the structure of the product was changed by the stoichiometry of isocyanide. This is also a rare result as a 2-selective functionalization of pyridine.
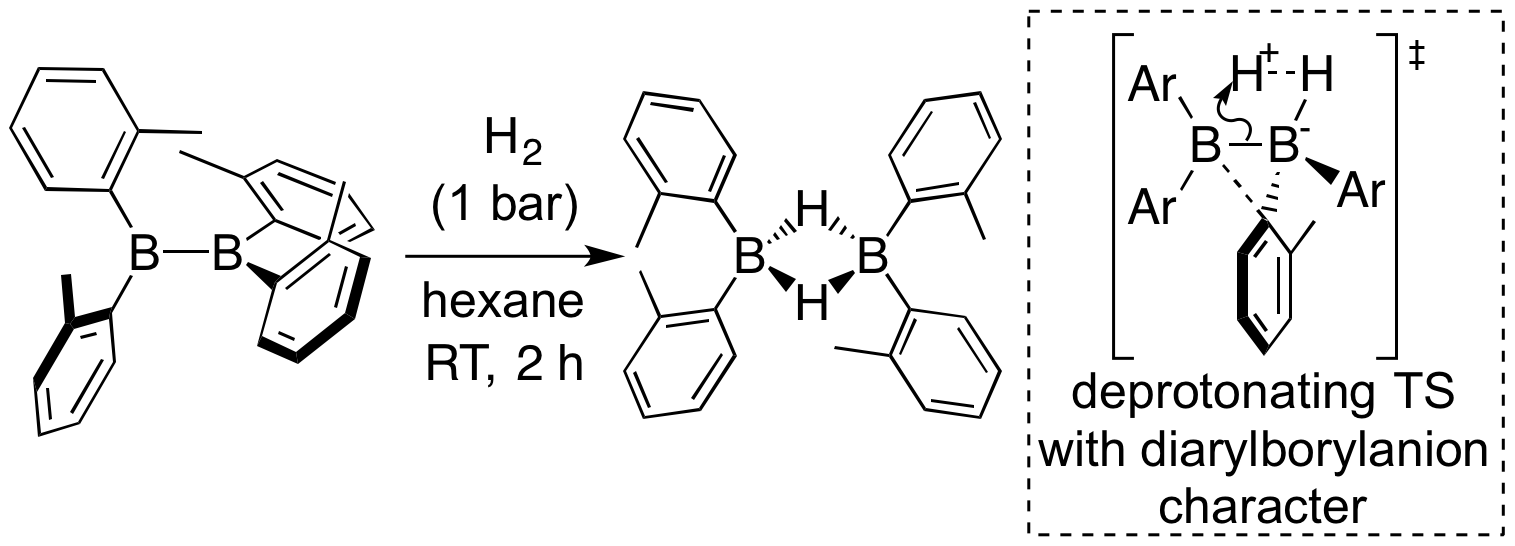
On the other hand, in our laboratory, we found that (o-tol)2B-B(o-tol)2 can be easily synthesized as diborane(4) having four carbon substituents. It was also revealed that this compound reacts directly with H2, even though it is an ordinary organoboron compound. DFT calculations revealed that this reaction proceeds via the transition state shown in the Figure. The electronic state of the transition state was analyzed to show that one boron atom receives the H-H bonding electron pair in H2 as Lewis acid and the other boron atom deprotonated H2 as a Brønsted base simultaneously. It can be said that this reaction proceeded due to the coexistence of high ly electron-accepting character of the overlapped empty orbitals of two boron atoms and high reactivity of B-B bonding electrons.
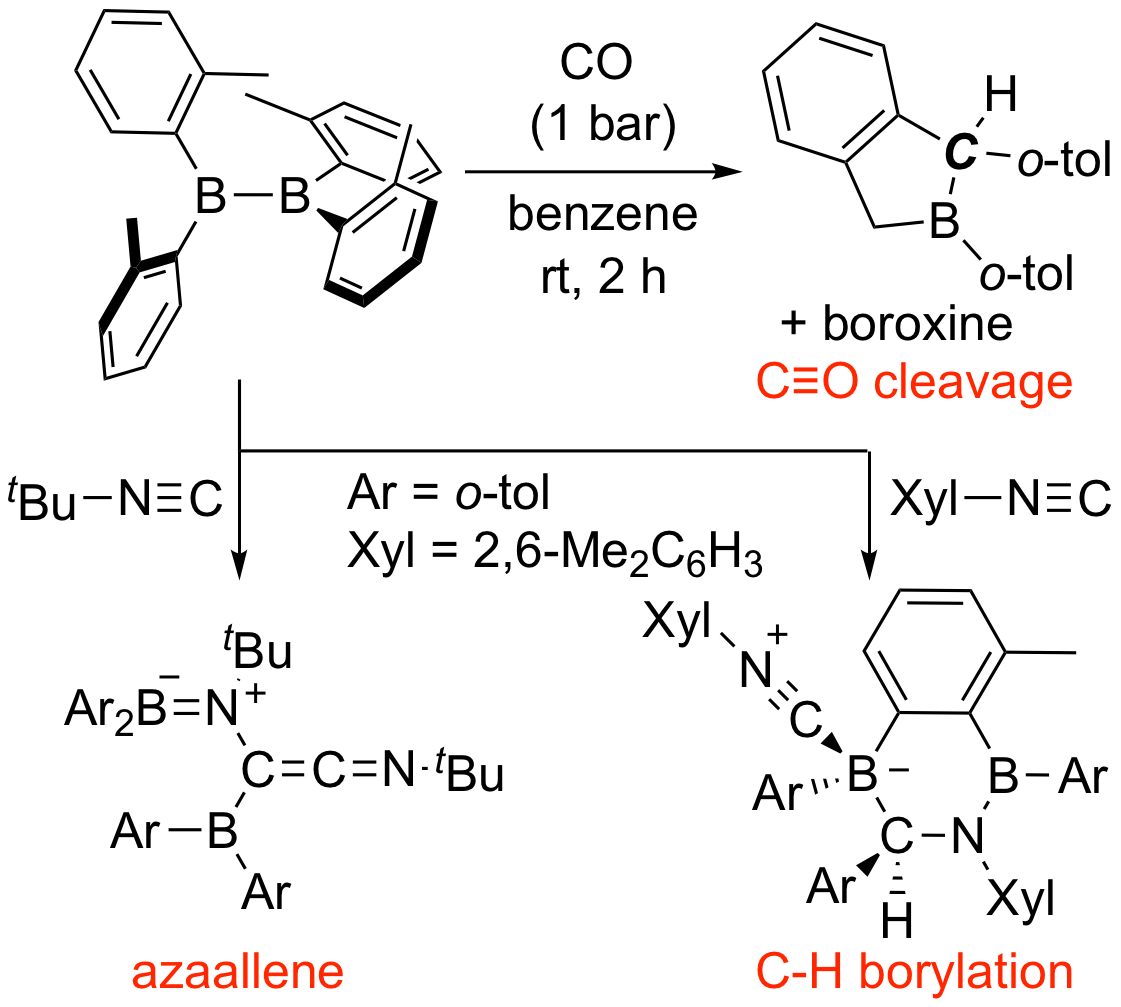
The (o-tol)2B-B(o-tol)2 was also found to react with carbon monoxide and isocyanide. In the former reaction, the C≡O triple bond of carbon monoxide is completely cleaved in one-step reaction, and the C—H bond of the methyl group on the benzene ring is also cleaved to form boraindane skeleton. In the latter reaction, depending on the type of substituent of isocyanide, an azaallene-type product in which two isocyanide molecules form a C-C bond, or a product in which the aromatic C-H bond of an o-tolyl group is cleaved is obtained.
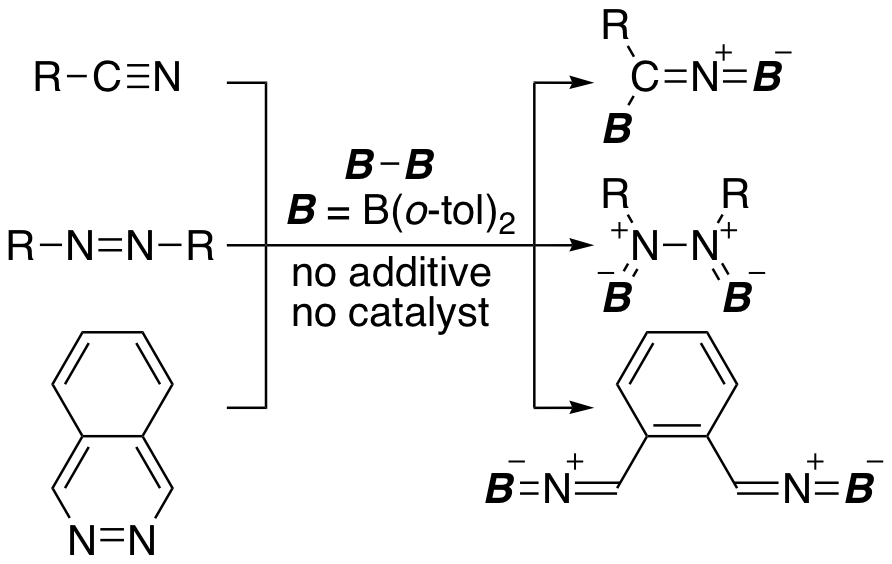
The C≡N triple bond of nitrile, the N=N double bond of azobenzene, and the N=N double bond of phthalazine also react with (o-tol)2B-B(o-tol)2. The reaction with nitrile and azobenzene gave a simple diborylation product, while the reaction with phthalazine gave a product with the N=N double bond cleaved. Isolated reaction intermediate, which is unstable at room temperature, was a simple addition product to the N=N double bond. It was also clarified that the N-N single bond of this intermediate was further cleaved with the aromatization of the upper six-membered ring as a driving force.
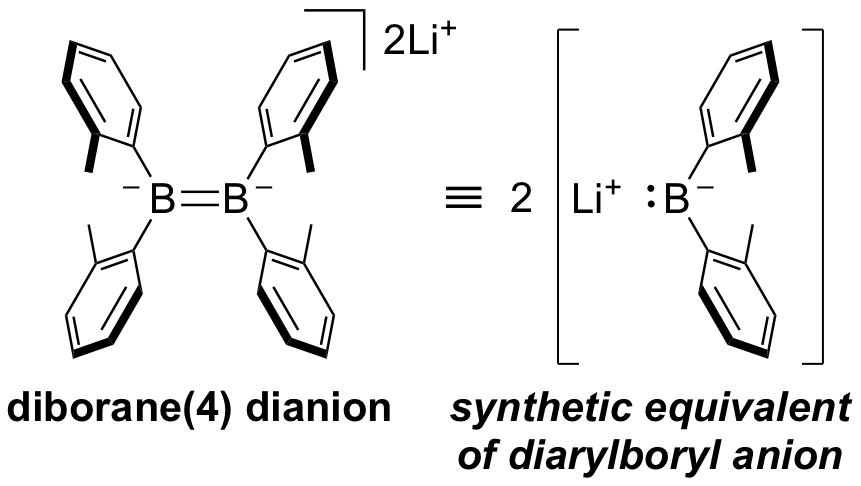
(o-tol)2B-B(o-tol)2 undergoes two-electron reduction with elemental Li or Mg to form the corresponding dianion with a B=B double bond. The structure and spectroscopic characteristics of this compound were similar to those previously reported, but their reactivity was notable. When the Li salt of this dianion reacted with dichloromethane, a diborylmethane, which can be regarded to form through double nucleophilic borylation on the carbon atom, was produced. A six-membered ring of B2S4 is formed in the reaction with elemental sulfur, and this reaction can also be regarded as being formed by a nucleophile attack on sulfur by a boron nucleophile. Although the reaction mechanism has not been clarified in either case, it was found that this dianion is a carbon-substituted boryl anion equivalent.
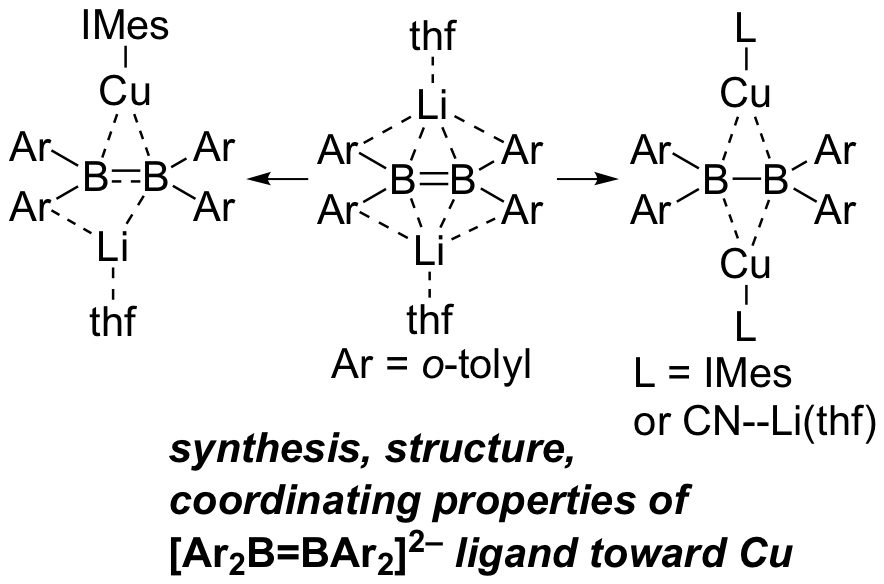
It was also found that the Li salt of diborane(4) dianion described above can be used as a ligand for transition metals. When this dianion was added to the copper(I) complex, mononuclear and binuclear copper complexes were obtained, depending on the stoichiometry of copper salt. The electronic states of these complexes are clarified by NMR spectra and UV-vis absorption spectra, and the electron density of copper atoms in this complex is higher than that of ordinary copper (I) complexes by XANES, reflecting the high electron donating property of the diborane(4) dianion ligand.
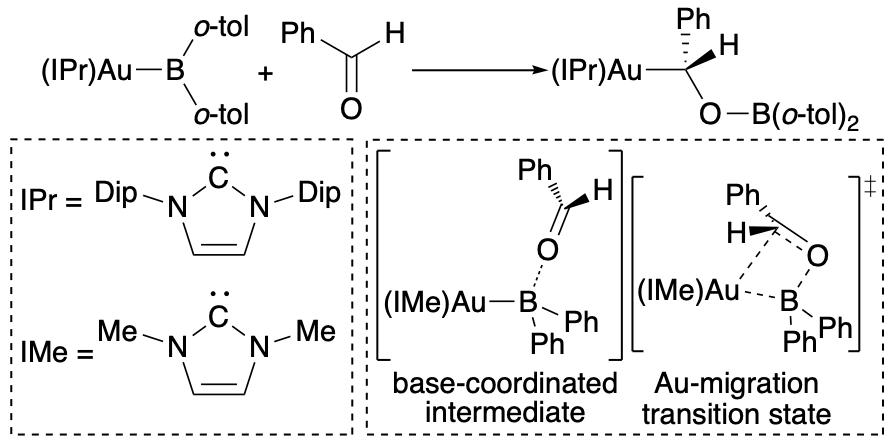
It is generally known that an X-type ligand covalently coordinated to a transition metal exhibits nucleophilicity. On the other hand, in this study, a carbon-substituted borylgold complex was synthesized by reacting the above-mentioned (o-tol)2B-B(o-tol)2 with a gold alkoxide complex. In the reaction of this borylgold complex with electrophiles, it was clarified that the gold atom behaves as a nucleophile and boryl ligand plays as an electrophile. It was found that this carbon-substituted gold boryl complex shows Lewis acidity on the boron atom, and that neutral ligands such as pyridine and DMAP are easily coordinated to the boron atom, and that this coordination raises the HOMO corresponding to the Au-B bond. On the other hand, in the reaction of this borylgold complex with carbonyl compounds and carbodiimides, the heteroatoms are coordinated to the boron atom and then the gold atom is nucleophilically transposed to the carbon atom, to give a boroxyalkylgold complexes were obtained. DFT calculations also confirmed that these reaction mechanisms were initiated by the coordination of Lewis base and that nucleophilic dislocations of gold atom proceeded.
related publications
(1) Asakawa, H.; Lee, K. H.; Lin, Z.; Yamashita, M.Nature Commun. 2014, 5, 4245. doi (open access) highlighted in Nature Japan (featured article)
(2) Asakawa, H. Lee, K. H.; Furukawa, K.; Lin, Z.; Yamashita, M. Chem. Eur. J. 2015, 21, 4267-4271. doi
(3) Kojima, C.; Lee, K.-H.; Lin, Z.; Yamashita, M. J. Am. Chem. Soc. 2016, 138, 6662-6669. doi
(4) Katsuma, Y.; Asakawa, H.; Lee, K.-H.; Lin, Z.; Yamashita, M. Organometallics 2016, 35, 2563-2566. doi
(5) Tsukahara, N.; Asakawa, H.; Lee, K.-H.; Lin, Z.; Yamashita, M. J. Am. Chem. Soc. 2017, 139, 2593-2596. doi
(6) Katsuma, Y.; Asakawa, H.; Yamashita, M. Chem. Sci. 2018, 9, 1301-1310. doi
(7) Katsuma, Y.; Tsukahara, N.; Wu, L.; Lin, Z.; Yamashita, M. Angew. Chem. Int. Ed. 2018, 57, 6109-6114. doi
(8) Katsuma, Y.; Wu, L.; Lin, Z.; Akiyama, S.; Yamashita, M. Angew. Chem. Int. Ed. 2019, 58, 317-321. doi
(9) Akiyama, S.; Yamada, K.; Yamashita, M. Angew. Chem. Int. Ed. 2019, 58, 11806-11810. doi
(10) Akiyama, S.; Ikemoto, S.; Muratsugu, S.; Tada, M.; Yamashita, M. Organometallics 2020, 39, 500-504. doi
(11) Akiyama, S.; Yamashita, M. Chem. Lett. 2020, 49, 721-723. doi
(12) Suzuki, A.; Guo, X.; Lin, Z.; Yamashita, M. Chem. Sci. 2021, 12, 917-928. doi
