2-12-1 Ookayama, Meguro-ku, Tokyo 152-8551, Japan
Department of Chemistry,
School of Science, Institute of Science Tokyo
Access to Ookayama campus, campus map
East 1 Building,
room 41, 44, 45

2-12-1 Ookayama, Meguro-ku, Tokyo 152-8551, Japan
Department of Chemistry,
School of Science, Institute of Science Tokyo
Access to Ookayama campus, campus map
East 1 Building,
room 41, 44, 45
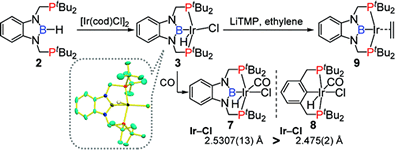
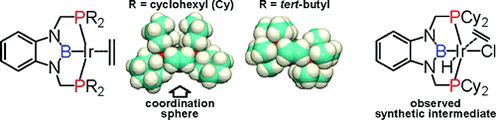
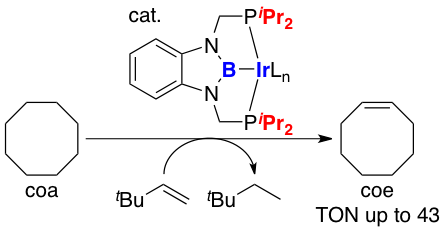
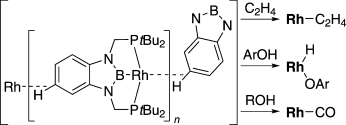
(2b-2) We also succeeded to synthesize PBP-Rh(H)(Cl) and PBP-Rh(H)(OTf) complexes by the same method for Ir complexes. Treatment of the latter complex with strong base led to a formation of T-shape, 14-electron Rh complexes as isolable solids. In the crystal, this complex has intermolecular C-H---Rh interaction to be stabilized with linear and polymeric structure. This T-shape complex rapidly reacted with phenol to give PBP-Rh(H)(OPh) complex through an oxidative addition of O-H bond. In the reaction of the T-shape complex with aliphatic 1° alcohols, PBP-Rh(CO) complex and linear alkanes were liberated. On the other hand, it was also found that the T-shape complex could react with cyclobutenones to cleave C-C single bond through an oxidative addition under mild condition. This is stark contrast to that the reaction of ClRh(PPh3)3complex required higher temperature and afforded a mixture of isomeric products.
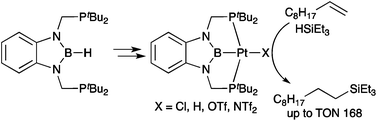
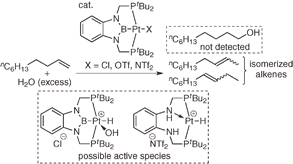
(2b-3) By a similar method, we also prepare PBP-Pt(Cl) complex and it could be converted to PBP-Pt(H) complex exhibited unusual 1H NMR chemical shift of the hydride ligand and Pt-H vibration in IR spectrum. Furtheremore, we also found that the former complex showed catalytic activity for hydrosilylation of alkenes and PBP-pincer ligand lost the central boron atom upon treatment with water or ethenol.
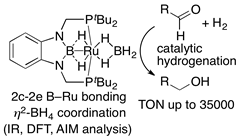
(2b-4) Reaction of hydroborane precursor with Ru(CO)3(cod) furnished PBP-Ru(H)(CO)2. Treatment of this complex with NMO lead to a formation of boronato complex, in which B-O bond of 1.329(6) Å was very short to have a double bond character. By using other Ru sources, PBP-Ru(Cl)(CO), PBP-Ru(CO)(η2-BH4), PBP(μ-H)2-Ru(OAc-κ2O), and PBP(μ-H)2-Ru(η2-BH4) complexes were obtained. Some of them were shown to have catalytic activity for hydrogenation of aldehydes by using hydrogen gas.
(2b-5) Synthesis of neopentyl-substituted PNP-pincer Ir complexes and elucidation of their reactivity
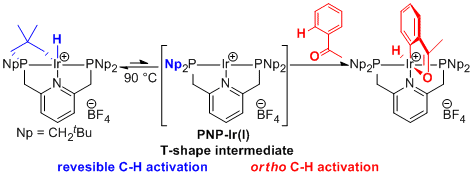
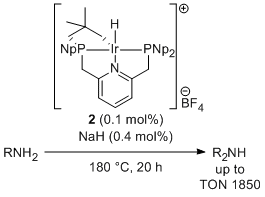
Iridium complexes having bulky group substituted pincer ligand are attractive because they could cleave C-H, N-H, O-H bonds. However, examples to utilize these bond cleavage reactions with pincer-ligated complexes are rare probably due to the steric crowding around the metal center. We recently developed a new pincer ligand system having neopentyl substituents on the phosphorus atoms to keep sufficient space around the metal center. Complexation of neopentyl-substituted PNP ligand with iridum easily gave an iridacycle complex via an CH activation at one of the neopentyl groups. This complex could react with arenes possessing a directing group and could catalyze a deaminative dimerization of alkylamines to afford dialkylamines with very high activity.
1) Segawa, Y.; Yamashita, M.; Nozaki, K. J. Am. Chem. Soc. 2009, 131, 9201-9203. doi.
2) Segawa, Y.; Yamashita, M.; Nozaki K. Organometallics 2009, 28, 6234-6242. doi
3) Hasegawa, M.; Segawa, Y.; Yamashita, M.; Nozaki, K. Angew. Chem. Int. Ed. 2012, 51, 6956-6960. doi
4) Ogawa, H.; Yamashita, M. Dalton Trans. 2013, 45, 625-629 doi
5) Masuda, Y.; Hasegawa, M.; Yamashita, M.; Nozaki, K.; Ishida, N.; Murakami, M. J. Am. Chem. Soc. 2013, 135, 7142–7145. doi
6) Miyada, T.; Yamashita, M. Organometallics 2013, 32, 5281–5284. doi
7) Ogawa, H.; Yamashita, M. Chem. Lett. 2014, 43, 664-666. doi
8) Miyada, T.; Kwan, E. H.; Yamashita, M. Organometallics 2014, 33, 6760-6770. doi
9) Tanoue, K.; Yamashita, M. Organometallics 2015, 34, 4011-4017. doi
10) Yano, T.; Moroe, Y.; Yamashita, M.; Nozaki, K. Chem. Lett. 2008, 37, 1300-1301. doi
11) Yamashita, M.; Moroe, Y.; Yano, T.; Nozaki, K. Inorg. Chim. Acta 2011, 363, 15-18. doi
